Publications
Ultrahigh specificity in a network of computationally designed protein-interaction pairs
Ravit Netzer, Dina Listov, Rosalie Lipsh and Sarel J. Fleishman
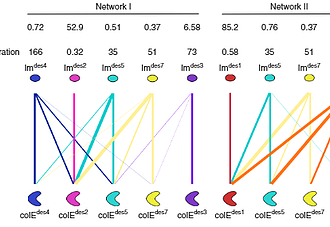
Protein networks in all organisms comprise homologous interacting pairs. In these networks,some proteins are specific, interacting with one or a few binding partners, whereas others are multispecific and bind a range of targets. We describe an algorithm that starts from an interacting pair and designs dozens of new pairs with diverse backbone conformations at the binding site as well as new binding orientations and sequences. Applied to a high-affinity bacterial pair, the algorithm results in 18 new ones, with cognate affinities from pico- to micromolar. Three pairs exhibit 3-5 orders of magnitude switch in specificity relative to the wild type, whereas others are multispecific, collectively forming a protein-interaction net-work. Crystallographic analysis confirms design accuracy, including in new backbones and polar interactions. Preorganized polar interaction networks are responsible for high specificity, thus defining design principles that can be applied to program synthetic cellular inter-action networks of desired affinity and specificity.
Automated Design of Efficient and Functionally Diverse Enzyme Repertoires
Olga Khersonsky, Rosalie Lipsh, Ziv Avizemer and Sarel J. Fleishman
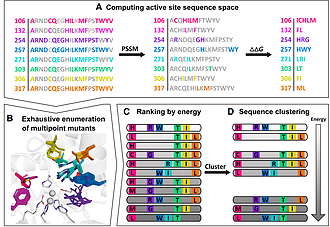
Substantial improvements in enzyme activity demand multiple mutations at spatially proximal positions in the active site. Such mutations, however, often exhibit unpredictable epistatic (non-additive) effects on activity. Here we describe FuncLib, an automated method for designing multipoint mutations at enzyme active sites using phylogenetic analysis and Rosetta design calculations. We applied FuncLib to two unrelated enzymes, a phosphotriesterase and an acetyl-CoA synthetase. All designs were active, and most showed activity profiles that significantly differed from the wild-type and from one another. Several dozen designs with only 3–6 active-site mutations exhibited 10- to 4,000-fold higher efficiencies with a range of alternative substrates, including hydrolysis of the toxic organophosphate nerve agents soman and cyclosarin and synthesis of butyryl-CoA. FuncLib is implemented as a web server (http://FuncLib.weizmann.ac.il); it circumvents iterative, high-throughput experimental screens and opens the way to designing highly efficient and diverse catalytic repertoires.
Highly active enzymes by automated combinatorial backbone assembly and sequence design
Gideon Lapidoth, Olga Khersonsky, Rosalie Lipsh, and Sarel Fleishman et al, Nature communications,2018
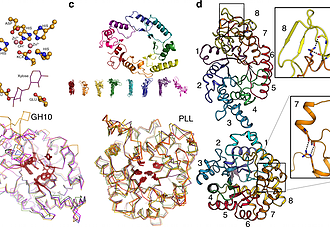
Automated design of enzymes with wild-type-like catalytic properties has been a longstanding but elusive goal. Here, we present a general, automated method for enzyme design through combinatorial backbone assembly. Starting from a set of homologous yet structurally diverse enzyme structures, the method assembles new backbone combinations and uses Rosetta to optimize the amino acid sequence, while conserving key catalytic residues. We apply this method to two unrelated enzyme families with TIM-barrel folds, glycoside hydrolase 10 (GH10) xylanases and phosphotriesterase-like lactonases (PLLs), designing 43 and 34 proteins, respectively. Twenty-one GH10 and seven PLL designs are active, including designs derived from templates with <25% sequence identity. Moreover, four designs are as active as natural enzymes in these families. Atomic accuracy in a high-activity GH10 design is further confirmed by crystallographic analysis. Thus, combinatorial-backbone assembly and design may be used to generate stable, active, and structurally diverse enzymes with altered selectivity or activity.
Principles of Protein Stability and Their Application in Computational Design
Adi Goldenzweig and Sarel Fleishman,Annual Review of Biochemistry,2018
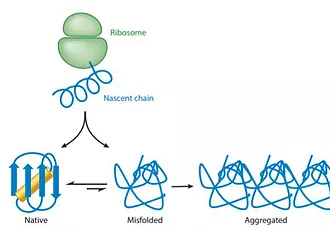
Proteins are increasingly used in basic and applied biomedical research.Many proteins, however, are only marginally stable and can be expressed in limited amounts, thus hampering research and applications. Research has revealed the thermodynamic, cellular, and evolutionary principles and mechanisms that underlie marginal stability. With this growing understanding, computational stability design methods have advanced over the past two decades starting from methods that selectively addressed only some aspects of marginal stability. Current methods are more general and, by combining phylogenetic analysis with atomistic design, have shown drastic improvements in solubility, thermal stability, and aggregation resistance while maintaining the protein’s primary molecular activity. Stability design is opening the way to rational engineering of improved enzymes, therapeutics, and vaccines and to the application of protein design methodology to large proteins and molecular activities that have proven challenging in the past.