Why design new protein function?
Nature provides us with myriads of examples of exquisitely selective proteins that function as binders and enzymes. Most proteins, however, are often formidably complex. For instance, a typical enzyme or the antigen-binding domain of an antibody comprise more than 200 amino acids and fold into complex three-dimensional shapes that depend critically on thousands of atomic interactions. By changing the protein sequence and structure or designing completely new proteins, we may generate desirable new enzymes for green chemistry, binders for research, diagnostics and therapeutics, and exquisite molecular sensors to measure metabolite concentrations. Designing proteins with new or enhanced properties is therefore a fundamental challenge of immense importance.
Further reading:
- Fleishman, S. J.; Baker, D. Role of the Biomolecular Energy Gap in Protein Design, Structure, and Evolution. Cell 2012, 149 (2), 262–273.
Evolution guided atomistic design - a new paradigm for design of function
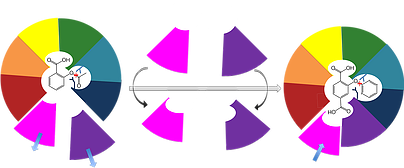
Our lab's long-term goal is to enable reliable and completely computational design of efficient, selective, and stable protein binders and enzymes. To achieve this goal, we are developing a unique strategy called evolution-guided atomistic design that uses information encoded in the evolutionary history of protein families to infer what structure and sequence features are likely to be tolerated in any given protein. We then use these rules to guide Rosetta atomistic design calculations in the search for new proteins with desired functions. To test our algorithms, we design new proteins that don't exist in nature and carry out wet-lab experiments either in-house or with our collaborators. Feedback from these experiments then enables the development of more sophisticated design algorithms. We therefore combine cutting-edge computational methods development with high-throughput experimental screening and stringent biochemical and structural analysis of designed proteins.
Further reading:
- Khersonsky, O.; Fleishman, S. J. Why Reinvent the Wheel? Building New Proteins Based on Ready-Made Parts. Protein Sci. 2016, 25 (7), 1179–1187.
- Goldenzweig, A.; Fleishman, S. J. Principles of Protein Stability and Their Application in Computational Design. Annu. Rev. Biochem. 2018, 87, 105–129.
Automated protein optimisation
Proteins in nature are subject to evolutionary processes that optimise their activities. In recent decades, scientists have emulated natural evolutionary process in the lab, enabling the optimisation of proteins for human needs, thus generating efficient enzymes and binders. But evolution is an iterative process in which every change in a protein (mutation) must result in one that is at least as functional as its predecessor or the variant would be purged by the powerful forces of selection. Thus, lab evolution experiments may take years of tedious trial-and-error. We developed several methods that enable rapid, one-shot optimisation of protein activities, generating enzymes that degrade a broad spectrum of highly toxic nerve agents, antibodies with much improved affinity and stability, and even a much cheaper and more stable variant of a protein that is the prime candidate to serve as a vaccine for malaria. One of the most important goals for the lab is to enable broad use of our algorithms by biochemists and protein engineers, and we therefore develop web servers that allow researchers around the world to customise our design protocols for their particular needs. The web servers carry out the calculations on our lab's computer cluster and return models of improved binders and enzymes by email. You're most welcome to try these web servers yourself!
Further reading:
- Goldenzweig, A.; Goldsmith, M.; Hill, S. E.; Gertman, O.; Laurino, P.; Ashani, Y.; Dym, O.; Unger, T.; Albeck, S.; Prilusky, J.; Lieberman, R. L.; Aharoni, A.; Silman, I.; Sussman, J. L.; Tawfik, D. S.; Fleishman, S. J. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63 (2), 337–346.
- Campeotto, I.; Goldenzweig, A.; Davey, J.; Barfod, L.; Marshall, J. M.; Silk, S. E.; Wright, K. E.; Draper, S. J.; Higgins, M. K.; Fleishman, S. J. One-Step Design of a Stable Variant of the Malaria Invasion Protein RH5 for Use as a Vaccine Immunogen. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (5), 998–1002.
- Khersonsky, O.; Lipsh, R.; Avizemer, Z.; Ashani, Y.; Goldsmith, M.; Leader, H.; Dym, O.; Rogotner, S.; Trudeau, D. L.; Prilusky, J.; Amengual-Rigo, P.; Guallar, V.; Tawfik, D. S.; Fleishman, S. J. Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72 (1), 178–186.e5.
- Goldenzweig, A.; Fleishman, S. J. Principles of Protein Stability and Their Application in Computational Design. Annu. Rev. Biochem. 2018, 87, 105–129.
Designing new protein structures
Control over protein activity demands control over the protein backbone structure, but the backbone has numerous degrees of freedom and design of new backbones in protein active sites has been a notoriously difficult problem. By combining information from naturally occurring structures and sequences with atomistic design calculations, we developed a new approach for backbone design in active sites. Initially, we implemented this strategy to design new antibodies, and therefore called the method AbDesign. Encouraged by the method's success in designing atomically accurate new antibodies with over 50 mutations from any naturally occurring antibody, we next applied this method for the design of high-efficiency new enzymes and a large network of interacting pairs of proteins that exhibited ultrahigh specificity binding. Thus, evolution-guided atomistic design provides exquisite control over protein structure, stability, and activity.
Further reading:
- Baran, D.; Pszolla, M. G.; Lapidoth, G. D.; Norn, C.; Dym, O.; Unger, T.; Albeck, S.; Tyka, M. D.; Fleishman, S. J. Principles for Computational Design of Binding Antibodies. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (41), 10900–10905.
- Lapidoth, G.; Khersonsky, O.; Lipsh, R.; Dym, O.; Albeck, S.; Rogotner, S.; Fleishman, S. J. Highly Active Enzymes by Automated Combinatorial Backbone Assembly and Sequence Design. Nat. Commun. 2018, 9 (1), 2780.
- Netzer, R.; Listov, D.; Lipsh, R.; Dym, O.; Albeck, S.; Knop, O.; Kleanthous, C.; Fleishman, S. J. Ultrahigh Specificity in a Network of Computationally Designed Protein-Interaction Pairs. Nat. Commun. 2018, 9 (1), 5286.
Where are we headed?
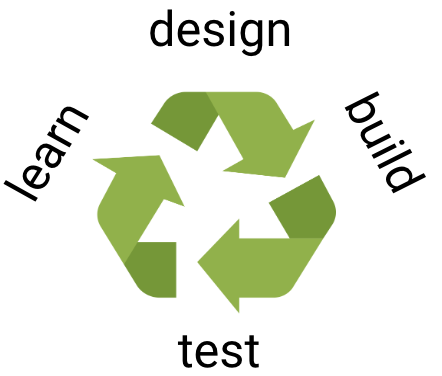
The holy grail of our field is to enable the complete computational design of any arbitrarily chosen biomolecular activity. To enable such template-free design of function, we still need to learn a lot more about how function to encode functions in proteins. We are therefore developing methods to design not a handful of binders or enzymes as in all current protein design methods, but vast repertoires comprising millions of substantially different variants. We use high-throughput screening methods to isolate the functional designs and deep sequencing analysis to fully characterise these designs. Next, advanced machine-learning methods are trained to find molecular features that discriminate the best designs from the rest, and these features are then used to improve the design algorithms, leading to a continuous, unbiased and systematic approach to learn the rules for designing new biomolecular activities. We are applying this strategy to the design of new hydrolytic enzymes, single-domain camelid antibodies, small-molecule binders, and a membrane transporter.